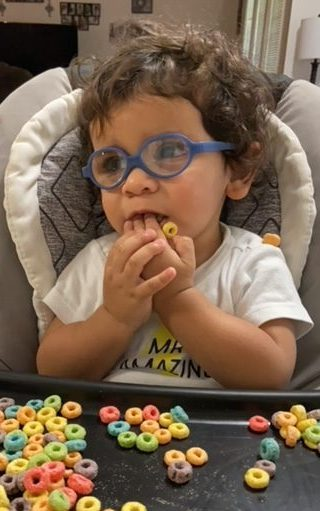
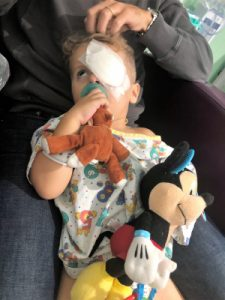
Desirae Potts breaks into tears when she recalls the first time a doctor said her infant son James had a disease she’d never heard of – Leber congenital amaurosis, known as LCA.
“I had no earthly idea what she was talking about. I left that appointment, I remember feeling so confused and so sad,” the 33-year-old mom said.
The doctor didn’t or couldn’t give Desirae and her husband, Robert, any information about LCA because the doc didn’t know a thing about the rare inherited retinal disease.
A smiling James Potts
“I had to grieve the life that my son would have had,” she said. “But I also wish that I knew then what I know now, that is: Whether he could see or not, I have a bright light!
“He’s so amazing, I couldn’t imagine him any other way and I wouldn’t change it if I could.”
She thought about the best way to bring up her son.
“I quickly learned the best way to raise him would be to raise him like my sighted children. He is the most resilient and bright young man. I know he is so smart. He amazes me every day.”
Mom becomes nurse to better help her son
James was 10 months old when his parents learned their younger son’s genetic diagnosis: LCA1* caused by a mutation in his GUCY2D gene.
About the same time, Desirae kept listening to doctors talking over her head and decided to become a nurse.
“I wanted to help him with my knowledge. It definitely helps because I have more medical knowledge than I did before. I understand what the doctor is saying. The doctors used all these words I didn’t know, and I was scared that I didn’t know.”
She began nursing school in James’ first year and earned her nursing degree about a year and a half ago. James is now 4.
“I’m not as worried now, and I know what to do in certain situations.”
James goes to school for the full day in an integrated classroom at the same school as his older brother, 9-year-old Robert. His older sister, Ariona, is in high school.
Their mom is the school nurse at the four middle schools in the district of their South Texas coastal city.
Keeps informed of LCA1 GUCY2D research
Desirae knew something wasn’t right with her baby’s vision at 2 months when he didn’t track objects or light. At 4 months, an ophthalmologist prescribed glasses, but they didn’t help.
James wearing his “Mr. Amazing” shirt
After the initial retinal disease diagnosis, James received his confirmed genetic diagnosis 6 months later at Baylor College of Medicine by Richard A. Lewis, MD, MS, Professor of Molecular and Human Genetics at the Houston medical school.
Annually, James visits John T. Stout, MD, PhD, who keeps the family informed of research in the LCA1 GUCY2D space. Dr. Stout specializes in retina/macular and retinal vascular diseases at Texas Children’s Hospital. His current research projects include human gene and stem cell therapy for proliferative and inherited ocular disease, retinal disease genotype-phenotype correlation, and intraocular angiogenesis, the formation of new blood vessels from the existing vascular tree.
LCA accounts for 5 percent of all retinal dystrophies and 20 percent of blindness in school-age children.
James received early-intervention therapy, and orientation and mobility therapy during his first years. He also sees a speech therapist at school and a developmental specialist in town.
Right now, his favorite food is bagels and butter. And he absolutely loves music and Daniel the Tiger, a spinoff from Mr. Rogers Neighborhood, where he sings songs about learning, and growing, and what to do with feelings.
James does not yet have words to describe his sight, which makes it hard to explain what he can see, although his mom believes he has light perception.
James’ vision is a family affair
“It not only affected him, but it also affected all of us,” Desirae said. “We all had to adjust and learn how to make things in a way that he could do things on his own and foster his independence.”
The Potts Family
Ariona, James’ 17-year-old sister, nurtures her little brother, with mom adding, “She’s the closest thing to me he can get. She’s very, very helpful.”
Robert was 5 when James was born.
“He didn’t quite understand what’s going on and how to be accommodating to his brother. We definitely had to inform him.”
Desirae is concerned about James pressing or rubbing his eyes, a symptom of LCA called oculodigital reflex. While the doctor said the reflex is normal, she is worried the action may cause harm. She tries to keep him occupied.
“I try very hard, but James is maybe the most headstrong person I have ever met in my entire life. If you tell him to do something, he will do the complete opposite. So, I tell him, ‘Don’t let me see your eyes,’ even though I don’t want him to defy me.”
James loves to explore and see with his hands.
“We don’t use the word ‘blind.’ We prefer to say that he sees with his hands. We don’t say ‘blind’ or ‘visually impaired.’ We do not want him to think that that would define him.
“That was something I had to teach both of our children, my husband, and all of our family. Some relatives are still in denial. We don’t put him in a box. We let him explore and have the same toys as his brother and sister,” she said.
“We try to have everyone stay on the same page to help build his confidence. Like it’s normal for him that he sees with his hands.”
One woman called doctor after doctor, only to hear they would not treat her and her two rare pediatric conditions because she turned 18 and no longer qualified for help.
Another fights for life-saving medicine to treat her rare disease that causes dangerous swelling.
While another faces $60,000 in annual medical expenses for her two teenagers living with a rare metabolic disorder requiring a special diet to stave off life-threatening symptoms.
“The medicine is there but you can’t get to it,” Candice Flewharty told the group gathered in Hartford, Connecticut, for Rare Disease Day. “Each phone call I make is a battle for my childs’ lives.”
State Sen. Cathy Osten, who has proposed legislation requiring insurance coverage for medical foods, accompanied Candice and her daughter to the gathering.
Candice Flewharty (L) with State Senator Cathy Osten
People living with rare diseases told their stories to Connecticut legislators on Rare Disease Day on March 23, about a month after a New England snowstorm canceled the event usually celebrated the last day of February.
Through the state’s newly minted Rare Disease Advisory Council, known as the RDAC, legislators of Connecticut’s General Assembly now have a collaborative and organized way to improve the lives of residents living with rare diseases.
The council’s first report of its findings and recommendations is due in November. Click here for a summary of the RDAC legislation, effective last July.
Lesley Bennett, CT-RAN State Volunteer and new RDAC member
The council will give patients, families, caregivers, health care providers, advocates, researchers, and other stakeholders the opportunity to make formal recommendations to state agencies and the legislature on ways to develop policy and health care legislation to improve the lives of those people living with rare disease and their caregivers, according to Lesley Bennett, Volunteer Ambassador for the Connecticut Rare Action Network (CT-RAN) of the National Organization for Rare Disorders (NORD).
Connecticut Gov. Ned Lamont signed legislation establishing the council after rare disease advocates worked for years to get it going.
“We did it,” Lesley said. “It took eight years, but we did it.”
Introducing six new council appointees
Rare Disease Advisory Council members will include Insurance, Public Health, and Social Services commissioners, or their designees, and 10 members appointed by the Governor and the Public Health Committee leadership. Six new members were introduced at the Rare Disease Day event; four have yet to be announced.
James Rawlings, President/CEO Sickle Cell Disease Association of America Connecticut Chapter, and new RDAC member
The six members and their council roles are:
Patient Advocate Representative: Lesley Bennett, RDAC Coalition Leader and CT-RAN Volunteer State Ambassador.
Pediatric Patient Representative: Saurabh Vaidya, Connecticut Hemophilia Society President, and father of a son with hemophilia.
Caregiver Representative: Mary Caruso, Friedreich’s Ataxia Research Alliance Founding Member, and caregiver for two adult children living with the disease.
Researcher: Joanna Gell, MD, Pediatric Oncologist and Hematologist, Connecticut Children’s, and Research Scientist, Jackson Laboratories.
Physician Treating Rare Disease Patients: Emily Germain-Lee, MD, Division Head, Pediatric Endocrinology & Diabetes; Director, Center for Rare Bone Disorders, Albright Center, and Osteogenesis Imperfecta Center.
Patient Representative: James Rawlings, R.PH, MPH, President/CEO Sickle Cell Disease Association of America, Connecticut, Michelle’s House.
“Everybody is here to help each other out.”
State Senator Saud Anwar
Connecticut Public Health Committee Co-Chair Sen. Saud Anwar told the gathering the time is here to find solutions to take care of and invest in the health of the rare disease population, saying the council must go forward with this mindset:
“Every illness is treatable. Every disease has an answer and a solution.”
The senator spoke about the realities of national and international collaborations resulting in developing regenerative medicine.
“We will put our hearts together a make sure we come out of the room with solutions. Everybody is here to help each other out.”
Connecticut Public Health Committee Co-Chair Rep. Cristin McCarthy Vahey told the group, “I look forward to working with you and being supportive in making things happen for all of you.”
State Representative Cristin McCarthy Vahey
Fewer than 40 treatments existed for rare disease 40 years ago; the number is now 600, according to Annissa Reed, NORD’s Associate Director of State Policy. She said she hopes the collaborative effort makes the dream of finding more rare disease solutions and access to medicine a reality.
Connecticut-based Hope in Focus advocated throughout the years with the Rare Action Network to help create the council, with Co-Founder and President Laura Manfre attending the Rare Disease Day event.
Hope in Focus’ Laura Manfre with Brian Rosen of Axion Pharmaceuticals
Connecticut joins 24 states in establishing a council specifically to address the complexities of living with a rare disease, caring for someone with a rare disease, gaining access to treatment, and getting better insurance coverage. To find out whether your state has an RDAC or is developing one, please go to: https://rarediseases.org/rare-disease-advisory-councils/map/.
The council is critical, as it is exponentially more difficult for the rare disease population – inherently fewer in number than the rest of Connecticut – to have a voice on the state level.
A rare disease in the United States is characterized as any disease, disorder, illness, or condition affecting fewer than 200,000 people. With more than 7,000 known rare diseases, upwards of 90 percent have no FDA-approved treatment. About 1 in 10 people – more than 30 million Americans – live with a rare disease.
Most rare diseases are genetic or have a genetic component, more than half of those affected by rare diseases are children, and all pediatric cancers are rare.
Dominic Cotton, a rare disease parent and co-leader of the RDAC Coalition, thanked the Department of Public Health and its nearly 60 years of support for newborn screening.
“Without newborn screening, my son wouldn’t be alive today.”
Kristen Angell and Jennifer Huron of the National Organization for Rare Disorders (NORD)
Every newborn in Connecticut is assessed for a range of diseases, and each year more than a hundred babies tested for any one of the diseases are on a critical, early-intervention path leading to keeping a disease in check and people living fulfilled lives.
Kristen Angell, NORD’s Associate Director of Patient Advocacy, said the gathering is important for our population living with rare diseases.
“It gives them an opportunity to speak face to face with our General Assembly and let them know the daily struggles and challenges they face, and it provides the legislators an opportunity to assist in making a possible impact.”
Access to medication a matter of life or death
Rare Disease Day is all about awareness. Here is a sampling of what legislators learned from people living with rare diseases:
Pamela Johnson and her 10-year-old son have a rare, life-threatening disease called Hereditary Angioedema (HAE), a genetic condition that can cause severe swelling in various parts of the body and affects about 1 in 50,000 people globally. She has a well-paying job and feels she should be able to afford to pay for her medications, but they cost $90,000 a month.
With no access to life-saving drugs in 2017, she underwent two surgeries for severe swelling in her throat. Without medication, two or three times monthly, she would experience attacks making it extremely difficult to breathe.
Pamela Johnson
David Leeds, who helped with the day’s introductions and has HAE, highlighted one aspect of life more difficult with a rare disease – getting treatment at a hospital. It is a place of stress, anxiety, and fear of not receiving the treatment he needs because doctors and nurses never encountered his disease.
He described two published investigations of major insurance companies denying coverage for medication to help people live. Insurance companies, rather than treating doctors, are deciding who gets to have medication and who doesn’t.
“This is every day for rare disease patients.” he said. “Insurance companies cannot be relied upon to determine what is medically necessary.”
***
Megan Freeman said no one should ever have to go through what a person living with a rare disease has to experience.
As if it were yesterday, she remembers the day she got her diagnosis, and her response:
Alissa DeJonge (L), Megan Freeman, and her friend Elizabeth Nagle
“Am I going to die? Am I going to live? Am I going to be able to live to get married and live to have kids.”
Megan lives with 2q37 deletion syndrome. She is one of about a hundred people worldwide with the ultra-rare chromosome disease that can affect many parts of the body. She is an advocate and founder of her own rare disease organization. She praised the legislators for creating the RDAC, saying, “I give you guys an A for effort!”
***
Rachel O’Grady was diagnosed as an adult with two pediatric conditions: Tethered Cord Syndrome, a neurologic disorder caused by tissue attachments limiting spinal cord movement, and Spina Bifida Occulta that causes a small gap in the spine. Any resources for Rachel in Connecticut dried up when she turned 18, no longer qualifying for help as a minor.
“They would not see me, nor would they treat me,” she said. Maxed out in medical debt, she found help in Massachusetts.
***
Alissa DeJonge’s sixth-grade son lives with a bleeding disorder called Hemophilia A. She knows people living with rare diseases make tough choices, given the huge cost of necessary medicines, and looks forward to having the new council work to protect patient care, help caregivers, and allow parents to take time off from work.
***
Kelly Considine, with her mother, Susan, and service dog, Gunner, in the atrium of the Legislative Office Building
Kelly Considine, accompanied by her mother, Susan, came to raise awareness about and promote research for a rare chronic pain disorder she lives with called Complex Regulatory Pain Syndrome.
Kelly characterized the disease as the most painful condition known to modern medicine. She receives some relief from an implanted therapeutic device and receives help and love, from her mother and her service dog, a golden retriever named Gunner.
***
Amy LaChance, the mother and caregiver of a child with Syngap1, a genetic mutation affecting 1,200 people worldwide, said people need access to genetic testing to support research and trials for a treatment to fix this genetic typo.
It makes a difference when people with rare disease tell lawmakers their stories about needing access to tests and medicine, saying, “The squeaky wheel really does get the grease.”
Biotechnology growth important to state economy
Paul Pescatello, JD, PhD, is Senior Counsel and Executive Director of the Connecticut Bioscience Growth Council, a committee of the Connecticut Business and Industry Association’s biotech and biopharma members.
Amy LaChance
The bioscience council fosters collaboration with the life-science institutions of biotech and biopharma, and with the state to help grow this sector of Connecticut’s economy. Developing a new medicine costs about $2.7 billion and takes 12 to 15 years, he said.
“Essentially, it costs what it costs, no matter the size of the patient population,” he said. “That’s why it’s important to underscore how rare disease new medicine research typically leads to many insights and advances in drug development for larger patient populations.”
Keep telling your rare disease stories
Representative Vincent Candelora, house minority leader and rare disease champion, urged people to tell their stories to make people aware so lawmakers can help fashion policy to help them.
“If we don’t hear from you, it’s harder for us to do our job. It’s the true stories that tell us everything that’s going on.”
His advice echoed the encouragement we at Hope in Focus give to our LCA and IRD community – tell your stories to feel less isolated in your journey of living with a rare inherited retinal disease and to help advance research into treatments to improve vision or to halt vision loss.
The council will give patients, families, caregivers, health care providers, advocates, researchers, and other stakeholders the opportunity to make formal recommendations to state agencies and the legislature on developing policy and health care legislation to improve the lives people living with rare disease and their caregivers.
The council will deliver its first report of findings and recommendations in November. Please click here for a summary of the RDAC legislation that Gov. Ned Lamont signed into law. Connecticut joins 24 states having a Rare Disease Advisory Council. To see whether your state is working to create such a group, please check here.
RDAC members will include Insurance, Public Health, and Social Services commissioners, or their designees, and 10 members appointed by the governor and the Public Health Committee leadership. Of those 10 appointments, here are the six announced at Connecticut’s March 23, 2023, celebration of Rare Disease Day:
Patient Advocate Representative: Lesley Bennett, RDAC Coalition Leader, and CT-Rare Action Network Volunteer State Ambassador, National Organization for Rare Disorders.
Pediatric Patient Representative: Saurabh Vaidya, Connecticut Hemophilia Society President, and father of a son with Hemophilia.
Caregiver Representative: Mary Caruso, Friedreich’s Ataxia Research Alliance Founding Member, and caregiver for two adult children living with that rare disease.
Researcher: Joanna Gell, MD, Pediatric Oncologist and Hematologist, Connecticut Children’s Hospital, and Research Scientist, Jackson Laboratories.
Physician Treating Rare Disease Patients: Emily Germain-Lee, MD, Division Head, Pediatric Endocrinology and Diabetes, and Director, Center for Rare Bone Disorders, Albright Center, and Osteogenesis Imperfecta.
Patient Representative: James Rawlings, R.PH, MPH, President/CEO Sickle Cell Disease Association of America, Connecticut, Michelle’s House.
We’ll let you know when the rest of the appointments are official. They include representation of hospitals, the biopharmaceutical industry, and people living with a rare disease.
Jessi Crawford fancied the clarinet when she played in her middle school band, while classmate Ted Beaman favored the trombone and guitar. Never did they dream their love of music would manifest a half lifetime later with the birth of their youngest son.
Atlas mixing it up!
Music is everything to almost 2-year-old Atlas – he loves to sing, he loves to dance, and he loves all kinds of music.
Before the toddler developed his interest in music, his parents noticed something about his eye movements.
“Out of nowhere,” at one-and-a-half-months old, his mom said, he developed horizontal and vertical nystagmus, characterized by side-to-side and up-and-down rapid, repetitive, uncontrolled eye movements.
Atlas received his first pair of glasses at 3 months from Kellogg Eye Center in Ann Arbor, Michigan, about an hour drive from the family’s home in Toledo, Ohio.
A month later, doctors suspected Atlas had Leber congenital amaurosis (LCA), a rare genetic eye disorder in which the rods and the cones of the retina – the light-gathering cells – do not function properly.
Geneticists confirmed his genetic diagnosis as LCA6 caused by a mutation in his RPGRIP1 gene. LCA6 can be particularly devastating because of its rapid onset and progression.
“This took us completely by surprise,” Jessi said. “What do you mean, we both have this dysfunctional gene? What are you talking about?”
She was more than floored, especially because her oldest son, 11-year-old Brayden-Lee, has a rare, life-threatening form of autism and epilepsy. Brayden-Lee and Atlas also have a 7-year-old brother named Ronan.
Jessi found comfort and encouragement from the geneticist and the genetic counselor, but it took time to process the idea that her son’s vision would deteriorate.
Atlas and mom, Jessi
“You have all these stigmas around losing your vision being the worst thing on the planet, but I realized he could still have a happy life. He’s not dying, he’s just going to lose his vision – that perspective helped a lot.”
She’d already enlisted state and local resources to help Brayden-Lee, so she began searching for any available support and assistance for Atlas.
“I found him an O&M (Orientation and Mobility) specialist, a developmental specialist, and a vision specialist, so he wouldn’t fall behind in anything – movement, sensory output, anything.
“I was still really kind of sad and overwhelmed, and, of course, worried, because I never experienced anything like this before. My oldest son had occupational therapy, speech therapy, hospital visits galore, but I never dealt with anyone who couldn’t see.”
LCA6 RPGRIP1 preclinical research underway
Atlas turns 2 in May and he’s quite advanced, talking in 4-, 5-, 6-word sentences.
“He knows his shapes and he’s working on colors, which is hard because he sometimes blends colors.”
Atlas has no vision from the midline of his eye, down. He has vision above, for now, and cannot see close up.
Jessi and Ted’s little boy underwent eye muscle surgery in February to release his eyes from crossing, which has helped his vision.
Atlas
Jessi connected with Odylia Therapeutics, a biotech working on late-stage preclinical studies for a potential treatment for the RPGRIP1 mutation, and, hoping to help advance research, she authorized the company’s use of images taken of her son’s eyes during the surgery.
The Atlanta-based business is developing an investigational gene therapy to treat vision loss caused by LCA6 and is working with vector technology developed by Odylia Co-Founder Luk Vandenberghe, PhD. The research builds on data generated at Massachusetts Eye and Ear in the labs of Vandenberghe and Eric Pierce, MD, PhD, a physician and surgeon at Mass Eye.
Odylia hopes to begin trials in 2025 and is seeking partnership or philanthropic funding for the estimated $3.5 million development costs.
Jessi said she’d like to enroll Atlas in a clinical trial, just not the first one because she fears possible, yet undiscovered, side effects from experimental treatment.
Watching Mickey Mouse and Listening to Rock ‘n’ roll, Country, Celtic, and Native American
Atlas loves music, singing, and watching TV.
“He loves watching Mickey Mouse-anything. He stood at the mirror, pointed at his shirt in the mirror and said, “Mickey Mouse, blue shirt, mamma.”
A happy child who loves raisins and pizza, he reaches for things and gets his spoon or fork to his mouth, while getting food into it is another thing.
Jessi said Atlas has a tough time with the letter ‘f’ and says shork when he means fork.
Atlas enjoying the beach
“He tells me every day, fork and bowl, fork and bowl, or ‘shork a bow, mamma, shork a bow.’” Jessi said just thinking about it makes her laugh.
The 31-year-old characterized herself and her fiancée, 32-year-old Ted, as “both huge, huge, musical people,” beginning back in the school band, when they started dating in their teens and later went their separate ways, only to start dating again four years ago.
“The fact that Atlas likes music so much is better for us. He loves Disney music, also rock, country music and Celtic, and Native American.
“So, when I say we’re well rounded, we’re well rounded.”
The community of people living with Leber congenital amaurosis caused by mutations in the RDH12 gene moved closer to realizing the shared goal of establishing a clinical trial to find a treatment for the blinding disease.
More than 40 people gathered for the Second Global RDH12 Scientific Conference in Baltimore. The daylong meeting in November offered perspectives from clinicians, patients, parents, advocates, academia, regulators, and industry.
Silvia Cerolini, founder of Eyes on the Future, one of three organizations under the umbrella of the Global RDH12 Alliance, said the conference made tremendous progress toward a common goal: Designing a successful clinical trial to find a treatment to improve or stabilize vision in people living with RDH12. Current LCA13 RDH12 research is in preclinical stages, with the hope that the first clinical trials may start in 12 to 24 months.
Silvia Cerolini and her 9-year-old daughter, Vicky
As one woman living with RDH12 voiced during the conference: “Any quantity of vision is everything for us.”
This recent conference brought together an international group of scientific, medical, advocacy, and industry experts to identify and prioritize potential elements in designing a clinical trial and its key components: Outcome measures and endpoints.
Outcome measures are used to assess a patient’s vision. Performed at the start of a clinical trial, the measures provide baseline information that can be compared with the same outcome measures done after an intervention or treatment, determining progress and efficacy.
Participants in the Second Global RDH12 Scientific Conference
Endpoints are outcome measures selected as the most relevant and clinically meaningful measurements that can be measured objectively to determine whether the intervention being studied is beneficial.
Selecting the most appropriate and patient-relevant outcome measures as endpoints for each specific IRD is a big challenge for the entire field of inherited retinal dystrophies.
The heterogeneity (diverseness) of IRD phenotypes (characteristics) poses significant challenges to understanding disease pathology, predicting treatment benefit, and selecting outcome measures and endpoints for clinical trials. Challenges in recent clinical trials failing to demonstrate treatment efficacy show the need for innovation in trial design and outcome measures’ selection.
Organizers cited four key challenges unique to treating RDH12 mutations: (1) Severe impairment of retina/vision from early life; (2) Uncertain expectations of stabilization/preservation versus improvement/restoration of vision; (3) Heterogeneity of the RDH12 phenotypes (and IRDs in general); and (4) Slow expected pace of detectable changes after treatment.
Conference highlights included stories from people living with the disease and the constructive dialog among industry representatives, clinicians, scientists, and regulators, Coates said.
“Most significant were the RDH12 patients and parents sharing their experiences with progressive vision loss and their hopes for a treatment,” she said. “The day marked a meaningful step toward successful trials and, hopefully, will become a model as other gene-mutation studies progress to trials.”
For Maria Fiore, mother of 17-year-old Bella who lives with RDH12, the day brought hope.
“The input – from the academic side, opinions from the regulatory side, and the competitive nature of having multiple interests on the industry side – gives me a lot of hope that we’re getting closer to an RDH12 clinical trial,” said Fiore, a member of the Board of Directors for RDH12 Fund for Sight.
“We appear to be closer than ever, and events like the conference help to keep the motivation and drive to bring this to the finish line a real possibility.”
Bella and Vicky are part of the global RDH12 community that includes more than 200 families from 20 countries. Children and adults living with the rare inherited retinal disease hail from the United States, Europe, South America, China, India, and Russia.
The families communicate through Facebook and an email newsletter, with some local groups emerging to involve more people and remove language barriers.
The Global RDH12 Alliance organizes twice-yearly virtual community calls to review progress on clinical developments and discuss common challenges. Collectively, the three RDH12 organizations have raised more than $3.5 million since 2011 to advance research and find treatments.
Click here to see a video of people living with RDH12 introducing themselves.
Cerolini said it had been far from easy to wrestle all the participants down for one full day.
“But we came together as one big team to optimize the chances of trial success for our RDH12 community. There is still work to do to give Vicky and all our RDH12 community hope to see the world,” she said.
“It is not easy. But we are as close as ever to the first human clinical trials and we need to keep going. What we are learning about RDH12 can help the entire IRD field.”
The gathering watched a video of children and adults with RDH12 describing difficulties with sight loss and the hope that research will lead to a treatment.
Coates said people from several countries expressed their desires to have a chance to better their vision or at least maintain it.
“The heartfelt presentation reminded everyone in the room why we were there.”
Individuals and their families at the conference comprised a group of nine, sharing worldwide perspectives about trial design and outcome. They said they would accept stabilizing visual function as a “significant win” in the pursuit of a treatment.
Adult patients talked about the importance of keeping whatever light perception they have and shared stories of struggles in knowing where they are in their space, including the perils of bumping and bruising their foreheads when navigating spaces.
As one woman characterized the quest: “Any quantity of vision is everything for us.”
Their comments led to discussions with regulators and clinicians about the complexities of retinal function and the validity of certain endpoints and outcomes.
Clinicians and academics shared unpublished data on RDH12 natural history and thought-provoking perspectives from the latest IRD trials.
Tomas S. Aleman, MD, PhD, presented insight from an RDH12-associated IRD natural history study and the latest expectations for treatment. He talked about differences between the LCA13 RDH12 and LCA2 RPE65 mutations, and the structural and functional relationship in the RDH12 mutation that causes central vision loss resulting in blurring.
L-R: Debra A. Thompson, Jean Bennett, Maria Fiore, Allison Galloway, and Tomas S. Aleman
Despite functional and structural degradation of photoreceptors, he said, RDH12 gene therapy may have the potential to improve outcomes, even in adults, once certain toxins can be degraded.
Dr. Aleman is Professor of Ophthalmology and Director of the Hereditary Retinal Degeneration Clinics at the Scheie Eye Institute’s Perelman Center for Advanced Retinal and Ocular Therapeutics (CAROT) at the University of Pennsylvania.
***
Jean Bennett, MD, PhD, discussed outcomes and endpoints from studying the RPE65 gene mutation while developing voretigene neparvovec, known as LUXTURNA®, and whether they can be translated to RDH12 clinical trials.
The U.S. Food & Drug Administration (FDA) in 2017 approved LUXTURNA to help improve vision for people living with RPE65. The therapy remains the first and only approved ocular gene therapy and the only gene therapy treatment for any inherited disease in the United States.
The breakthrough treatment developed at Children’s Hospital of Philadelphia (CHOP) and Spark Therapeutics in Cambridge began with the successful treatment of a special being, Lancelot, the first in a line of Briard herding dogs, who helped drive research to bring the gene therapy to market.
Dr. Bennett told the group the development of a novel patient-relevant endpoint – a multi-luminance mobility test (MLMT) to measure functional vision, or how a person navigates in a vision-related activity across a range of light levels in daily living – was instrumental in demonstrating how voretigene neparvovec could improve vision in RPE65 patients, leading to LUXTURNA’s approval.
The navigational test, though, can be cumbersome and time-consuming, given the elaborate setup with lighting controls, videotaping, analysis, and such. Alternatively, researchers are working on virtual-reality tests, created as time-savers with more versatility. The task would be to design a functional vision test using a virtual-reality obstacle course.
Drs. Bart P. Leroy, Jean Bennett, and Daniel C. Chung
Dr. Bennett is Professor of Ophthalmology at CAROT and the F.M. Kirby Emeritus Professor of Ophthalmology at the Perelman School of Medicine at UPenn.
***
Dr. Robert E. MacLaren, Professor of Ophthalmology at the University of Oxford, discussed lessons drawn from recent IRD clinical trials and vector delivery. He is working on a trial assessing the effects of retinal gene therapy with an adeno-associated viral (AAV) vector encoding genome particles.
LUXTURNA gene therapy employs a human-engineered AAV vector containing copies of a normal gene injected under the retina to express a protein necessary for vision.
Prof. MacLaren discussed opening the door to a new concept in the IRD space where researchers look for statistically significant improvements that show the vector is working and be more flexible in the immediate argument of whether that has a clinically significant improvement in that patient’s visual function, there and then.
Professor Bart P. Leroy, MD, PhD, discussed information learned from recent IRD clinical trials and gene therapies in clinical practice.
He co-authored the PERCEIVE study that analyzed the Year 2 interim research on LUXTURNA, concluding the drug demonstrated safety and effectiveness when used in clinical practice following its regulatory approval. The study also identified a new adverse drug reaction called chorioretinal atrophy, causing inflammation in some patients. So far, the reaction has not affected visual function and will be tracked as the study progresses.
He emphasized working toward shared goals and communicating with regulatory agencies on design and outcomes for IRD clinical trials. Saying children are not little adults, Dr. Leroy said questions about specific designs and patient-reported outcomes need to be adapted to the study population.
Dr. Leroy is Professor of Ophthalmology, Ophthalmic Genetics, and Visual Electrophysiology at Ghent University, and Head of the Ophthalmology Department at Ghent University Hospital, Ghent, Belgium.
***
Adam M. Dubis, PhD, Associate Professor, University College of London Institute of Ophthalmology Global Business School for Health, and Advanced Human Retinal Imaging Specialist at Moorfields Eye Hospital, presented information on work planned around data-driven solutions for RDH12 clinical trial support.
Meira GTxCEO RobertK. Zeldin, MD, PhD, updated the group on the biotech’s RDH12 clinical development. The company is enrolling participants in the US, UK, and Europe for a natural history study and is planning a Phase 1/2 trial to assess safety and tolerability of potential gene therapy treatment.
Opus Genetics’ CEO Ben Yerxa, PhD, discussed capitalizing on key lessons in LUXTURNA’s development. Opus has three LCA-related projects in its pipeline, including one in early stages for RDH12. The company concentrates on preclinical projects in, or advancing toward, early-stage clinical trials to move research forward, with the goal of a successful trial or trials.
The FDA’s Dr. Wiley A. Chambers and the EMA’s Jane Moseley discussed trial-design parameters and outcome measures. They articulated their points of view, offering actionable advice on novel endpoints, including structural versus functional measures, virtual-reality mobility mazes, and patient-reported outcomes.
Todd A. Durham, Foundation Fighting Blindness Sr. VP of Clinical & Outcomes Research, talked about Patient-Reported Outcome Measures (PROMS), questionnaires designed to capture how a patient feels or functions, without input or interpretation from anyone else.
PROMS are developed using qualitative information, such as experiences, priorities, and contextual information from well-defined patient populations. Durham described ways the information could help advance research into treatments for people living with RDH12 and other IRDs, and he recommended steps toward creating a draft PROM strategy to begin conversations with regulators and payors.
Elin Haf Davies, PhD, CEO of Aparito, presented information on patient-centered clinical outcome assessments and the importance of quality of life for people living with rare diseases, such as RDH12.
Attendee Perspectives
The conference, chock-full of experts in myriad aspects of working to find a treatment for people living with RDH12, manifested hope and excitement toward its goal of launching clinical trials. Here are a few reflections from people in attendance:
Vicky, Silvia Cerolini’s daughter
“The level of engagement and willingness to share knowledge, experiences, and perspectives from this multi-stakeholder group was inspiring and motivating,” Astraea’s Sue Lacey said. “And I’m sure this will translate into increased momentum and robust clinical development programs for RDH12.”
From Prof. Bart P. Leroy: “A big thank you for organizing a day that took RDH12-IRD patients considerably closer to gene therapy. The maturity and respect during the day allowed for an open and fruitful discussion.”
Dr. Daniel C. Chung with SparingVision thanked organizers for putting together a great meeting and said, “The free exchange of thoughts and points of view was incredibly enriching. We look forward to continuing this conversation and developing concrete solutions for further development until one day a patient will have the option of a treatment.”
As general manager and vice president of Cardinal Honda in Groton, Conn., Kim Cardinal Piscatelli donates time and money to charities. But when she heard about Hope in Focus and the organization’s advocacy for people living with visual impairment, it struck a personal chord.
Piscatelli was a teenager when she watched her late grandmother slowly lose her vision to glaucoma.
“It was scary to watch her go through that,” Piscatelli recently recalled. “I loved her very much, and I saw her world get smaller and smaller.”
Employees blindfolded
Piscatelli and her family, including her sister, learned how to help her. They closed their eyes and folded laundry. They learned to present meals as if they were on a clock.
“The chicken is at 12 o’clock, and the coleslaw is at 3 o’clock.”
Hope in Focus’ Dinner in the Dark gala fundraiser, an annual culinary adventure that involves wearing a blindfold while eating food from a secret menu, immediately appealed to Piscatelli. She asked her sister to come with her.
“I was so excited,” she said. “As a child, I remember that the empathy I had for my grandmother would lead us to do things to learn to empathize with her.”
Though the concept brought back memories, it wasn’t exactly muscle memory. The two were eager to go, but a bit apprehensive.
“I remember holding hands under the table and being like, ‘I don’t know what is going to happen, but you are right here, right?’” Piscatelli said.
She loved it. Over the years, she’s brought her children, too. But she also has taken the concept back to Cardinal Honda. It started with a morning meeting four years ago. Employees wore blindfolds
“There’s a lot of conversation about diversity and inclusivity,” Piscatelli said. “It seemed like a good way to participate in that conversation.”
That conversation has grown significantly since then, as companies and individuals look to be more inclusive and empathetic to everyone. So, in 2022, the year Dinner in the Dark returned from a two-year pause due to COVID, Piscatelli resurfaced the exercise.
The group blindfolded.
It was a surprise twist on the typical Monday morning meeting — Piscatelli hadn’t announced her plan. Still, nine of the 10 employees put on blindfolds, while one took photos. When the blindfolds came off, people expressed similar sentiments to those Piscatelli felt during her first Dinner in the Dark.
“They reported they were paying more attention and not worried about what everyone else in the room was doing,” Piscatelli said. “Someone said they felt lonely…because you are not looking around the room making connections or eye contact.”
But even with the uneasiness, people showed interest in trying it again. When Piscatelli asked for six volunteers to put on blindfolds for lunch, the hands shot up.
They tried using tips Piscatelli learned at Dinner in the Dark from Sofia, daughter of Hope in Focus Co-Founder Laura Manfre, and a young woman living with LCA: Bring your food to your face, not your face to your food. Piscatelli announced the clock placement of the meal for the employees, just as she used to do for her grandmother.They huddled up after to discuss.
“Someone said it gave them a whole new perspective,” Piscatelli said. “They didn’t know if they had food on their shirts. They were more aware of what they were eating and how it tasted better, but said it was hard to eat. They lost track of what food was where on the plate.”
Employees sitting in a meeting with blindfolds on
Piscatelli hopes the exercise allows her employees to feel more empathy for people with vision loss and think of strategies to make the world more inclusive for them.
“It forced them to think about different sorts of things,” Piscatelli said. “Instead of changing what was on TV, we had to announce what was going to happen next. It helped people be prepared without visual tools.”
In a home with a person with vision loss, people can announce who they are and why they are coming into the room. For example, “It’s me, Kim, and I am here to do laundry.”
In the end, the staff learned something new and embraced leaving their comfort zones.
“It turned their day…it wasn’t a typical Monday,” Piscatelli said. “Everyone was more aware: What else do I not know?”
Piscatelli hopes other businesspeople feel inspired to learn what they don’t know about vision loss.
“It’s one thing to talk about equity, diversity, and inclusivity.” Piscatelli said. “It’s another thing to immerse everyone in an exercise where they feel empathy for people who have low vision and compassion for other people as they are temporarily trying that on.
Advocates for people living with rare diseases are collaborating with Connecticut Gov. Ned Lamont’s office and leaders of the Connecticut General Assembly’s Public Health Committee to build the state’s new Rare Disease Advisory Council (RDAC) that requires appointees, budgets, and bylaws be in place this summer.
Assistant Director of Advocacy for the National Organization for Rare Disorders (NORD) Kristin Angell joined Co-Ambassadors Katie Gillick and Lesley Bennett of NORD’s Connecticut Rare Action Network (CT-RAN) in hosting a Zoom conference with more than 30 people, including state legislators, patient advocates, doctors, researchers, industry leaders, health care providers, caregivers, and people living with rare disease.
The meeting included two representatives from Connecticut-based Hope in Focus and discussions about the range of needs to be addressed by the new council and the planning of next month’s celebration of Rare Disease Day on Feb. 28.
“We are working on it, and we will get it done,” Bennett said.
A rare disease is defined as one affecting fewer than 200,000 people nationwide. More than 7,000 known rare diseases affect an estimated one in 10 people in the United States, translating to about 30 million people or 10 percent of the country’s population. Globally, 300 million people live with rare diseases.
With the establishment of a permanent Connecticut RDAC, 13 members will be appointed to advise and make recommendations to the Department of Public Health, the Department of Social Services, and other state agencies about the needs of people living in Connecticut with a rare disease and their caregivers. Advisory councils may differ from state to state. Please click here to check whether your state has an RDAC or is working to establish one.
The cause of many rare diseases is unknown, but about 80 percent are genetic, such as LCA and other IRDs, and about half of all rare diseases affect children.
Getting state agencies to understand the rare disease population is key to the RDAC. Even something as simple as putting links to national rare disease resources on state government departments’ websites would be a big step.
More than 40 people submitted applications to become part of Connecticut’s council, and six have been appointed so far. The names of the new members will be announced when all 13 have been appointed.
The group will include insurance, public health, and social services commissioners, or their designees, and 10 members appointed by the Governor and the Public Health Committee leadership. Those 10 members will be:
a representative of an association of hospitals or a hospital administrator, and a physician with expertise in medical genetics
a representative of a patient advocacy group in the state representing all rare diseases, and a family member or caregiver of a pediatric patient living with a rare disease
a representative of the biopharmaceutical industry involved in rare disease research and therapy development, and an adult living with a rare disease
a member of the scientific community engaged in rare disease research, and a caregiver of a child or adult living with a rare disease
a physician who treats people living with a rare disease, and a representative, family member, or caregiver of a person living with a rare disease
Planning for Rare Disease Day
Following the midterm elections and changes in some legislative seats at Connecticut’s General Assembly, educating lawmakers is key to finding solutions for people living with rare disease to have the necessary access, resources, and educational tools to enjoy their best quality of life.
One person attending the meeting talked about the need for more doctors and patient advocates in Connecticut because people living with rare disease often must travel to another state for medical care. She also mentioned the potential of patients being charged for sending simple questions to doctors through computer health care portals, and whether lawmakers attending upcoming Rare Disease Day events could help mitigate or eliminate those costs.
The trial update includes safety and efficacy data from all 14 patients treated to date in the study comprised of 12 adults and two pediatric patients. EDIT-101 was tolerated with no serious ocular adverse events or dose-limiting toxicities observed. Most adverse events were mild and expected for subretinal delivery.
Along with showing improvement in BCVA, three of the 14 people demonstrated consistent improvements in two of the following three additional endpoints: Full-field sensitivity test (FST), visual function navigation, which means navigating a mobility course to assess mobility and functional vision in people with an inherited retinal disease such as LCA10, and visual function quality of life, information derived from the patient or caregiver while participating in the study.
Of those three people, two of them each had two identical versions of the genetic marker for the CEP290 IVS26 mutation, meaning they were homozygous for that mutation.
Because LCA10 patients homozygous for CEP290 IVS26 represent an estimated 300 people in the United States, and no other baseline characteristics were identified in the study’s dataset, the Editas statement said, “[T]he company will not progress this program independently and will seek to identify a collaboration partner to continue the development of EDIT-101.
“As a result, Editas Medicine is pausing further enrollment in the BRILLIANCE trial and will continue long-term follow-up of all patients who have been treated to date.”
Stay informed, stay connected
Hope in Focus keeps the LCA community informed of various trials focused on finding cures for any one of the more than 27 forms of LCA, including people living with LCA10 and associated research into developing therapies to correct mutations in the CEP290 gene. LCA10 is the most common form of the rare disease, affecting 20 to 30 percent of all LCA patients. LCA affects about 1 in 33,000 people worldwide.
Hope in Focus Co-Founder and Board Chair Laura Manfre asks anyone interested in learning more about the Editas trial to please send us an email at info@hopeinfocus.org, and we would be happy to facilitate a conversation.
“Science, business, and people are all key to getting a treatment from the lab to the people who need it. The announcement from Editas Medicine is a win for science, most certainly. Being able to go into the back of the eye and safely edit a genetic mutation is nothing short of a historic milestone,” Manfre said.
“The announcement isn’t entirely what we hoped for, however, as Editas has paused its LCA10 trial. More needs to be learned on the science side of things, there are business and regulatory issues to address, and as always, access to the patient community is critical.
“This journey was never going to be a straight line, and setbacks, although disappointing, are just proof we’re still moving. Stay informed, stay connected, and please make sure you’re registered and up-to-date in My Retina Tracker®.”
It will be three years this July when Editas began its CRISPER/Cas9-based trial of EDIT-101, administered through a subretinal injection to reach and deliver the gene-editing machinery directly to photoreceptor cells.
Researchers designed the BRILLANCE Phase 1/2 clinical trial of EDIT-101 to assess the safety, tolerability, and efficacy of the potential treatment. Patients received a single dose of EDIT-101 under the retina in one eye. They are monitored every three months for a year after dosing and less frequently for two more years. More details about the trial can be found at here under the reference NCT#03872479.
Atsena Therapeutics, a clinical-stage gene therapy company focused on reversing and preventing blindness, has an ongoing Phase 1/2 clinical trial evaluating a potential therapy for Leber congenital amaurosis 1 (LCA1) caused by mutations in the GUCY2D gene.
Kara Fick (L) and Shannon Boye (R) on Hope in Focus Let’s Chat About … series
We learned about this research from Kara Fick, head of Patient Advocacy and Medical Affairs at Atsena Therapeutics, and Atsena Founder and Director Shannon E. Boye, PhD, during our Oct. 27, 2022, webinar episode titled “Let’s Chat About…Atsena Therapeutics’ work in LCA.” Courtney Coates, Hope in Focus Director of Outreach and Development, moderated the session, which can be viewed here.
“Let’s Chat About…” is our free webinar series bringing together researchers, advocates, industry leaders, and people living with Leber congenital amaurosis (LCA) or other rare inherited retinal diseases (IRDs) for conversations important to the rare retinal disease community.
How did Atsena Therapeutics come to be?
Shannon and her husband, Sanford, met in grad school.
“It was a nerd romance,” she said.
Boye’s thesis involved developing viral vectors for retinal disease treatment, specifically for LCA1 (GUCY2D), and she became known for generating gene technology. She has authored more than 60 peer-reviewed manuscripts and multiple textbook chapters. She also has been actively involved in grant and manuscript review, and she has received several major awards.
Over several years, the couple worked with large, medium, and small pharmaceutical companies, but they grew frustrated at how long it took to bring developing therapies to patients.
“It was going very slowly and that was frustrating,” Boye said. “We saw business decisions overriding sound scientific decisions.”
Eventually, she sent a big long vent to Foundation for Fighting Blindness CEO Ben Yerxa, and he helped push her and her husband into starting their own company, Atsena Therapeutics. The couple co-founded the business, with Sanford Boye serving as Chief Technology Officer.
Kara Fick, who has been working as a patient advocate for rare diseases in the biotech world for nearly a decade, is passionate about bringing the patient voice, perspective, and expertise to the table.
“It’s pretty apparent, and you know from Atsena’s founding, that really keeping patients at the center and trying to move research forward so that it can get to patients is super important,” Fick said.
At Atsena, she strives to bridge the gap between the science of innovative therapies and the daily needs of individuals living with rare diseases. She also works to understand more clearly the barriers to diagnosis, treatment, and management of rare diseases and how to better address those hurdles with patients and clinicians.
What is gene therapy?
All of us have genes. They give us blue eyes or brown eyes, for instance. But our genes also make proteins. Proteins are the building blocks of life. They can act alone or in combination with other proteins to perform essential functions in our cells.
A good example of proteins important for vision are the proteins in our photoreceptors and retinas. Those proteins all work together to convert light into an electrochemical signal that is sent to the brain and processed as vision.
Genes make proteins, and proteins perform essential functions. But sometimes, we can have a misspelling in our genes — in other words, a mutation. Because of that misspelling or mutation, in some cases, the protein that gene was supposed to make did not form. In other cases, perhaps, the protein forms, but it’s misshapen and can’t interact properly with the other proteins. When that happens, the normal biological function of the protein is disrupted, for example, in your photoreceptors. As a result, your photoreceptors are unable to transmit light into a signal processed as vision.
The concept of gene therapy involves taking a healthy copy of a gene that lacks that misspelling and delivering that healthy version of the gene to the photoreceptor cells.
How is the LCA gene therapy trial going?
Drug development and gene therapy development take a long time, and there are a lot of processes that companies have to go through to ensure that anything given to patients is safe and effective.
Work to develop this gene therapy began in earnest in 2004 in Boye’s lab at the University of Florida. After pre-clinical studies in chickens and mice, they were ready for clinical trials in humans.
Atsena is working on a combination Phase 1/2 clinical trial for LCA1 (GUCY2D). In this phase of the trial, researchers test different gene therapy doses, find the best dose, and closely monitor the safety of the therapy.
“Because we’re doing a combo Phase 1/2, we’re looking primarily at safety, but we’re also incorporating some tests into the clinical trial that can help to tell us a little bit about the efficacy of the gene therapy,” Boye said.
‘Researchers began with adults aged 18 and up but recently opened it up to ages 6 and up. Fifteen people, all with GUCY2D mutations, are taking part in the trial, ranging in age from 12 to 76 years old.
So far, the trial is going well, with no participants reporting serious side effects related to the gene therapy. They did see two cases of inflammation, but both were mild and resolved after treatment.
Besides safety monitoring, Atsena has conducted several tests, including the Full-field Stimulus Test (FST). The test quantifies visual perception through flashes of varying luminance. Researchers saw significant improvements in the results of that test from individuals who received the highest dose of the gene therapy.
Participants receiving the gene therapy also showed clear improvement in a multi-luminance mobility test (MLMT), in which they navigated a course with obstacles of varying height and under different levels of illumination.
Participants also underwent a third test, called the BCVA or Best Corrected Visual Acuity, which measures everyday vision. (Think of the poster with different letters you’re asked to read at the eye doctor’s.) That test had more mixed results, with some patients seeing improvement and others not seeing much, if any, improvement.
What happens next?
This is still just the preliminary data. Atsena needs all 15 participants in its trial to get through their first full year after being treated with gene therapy. Then, they’ll collect all of the data, analyze it, and summarize it vigorously and thoroughly before moving on to the next phase.
At that point, they’ll need to meet with the Food and Drug Administration (FDA) and other regulatory agencies to get their agreement and permission to move forward with a Phase 3 trial. That trial will focus on efficacy and seeing how well the gene therapy works. It’ll still pay attention to safety, but the primary goal of Phase 3 is to focus on efficacy and use that as supportive data that could be submitted to the FDA and other agencies to get full approval of the gene therapy.
Why genetic testing is essential for clinical trials?
As more trials are starting and more gene-specific research is being done, it’s going to become more important to know precisely which genetic mutation may be causing your LCA.
“Getting genetic testing is essential for figuring out the specific genetic mutation that may be causing your LCA,” says Boye.
If you have not been genetically tested, talk to Hope in Focus. There’s no cost to patients and family members to receive genetic testing.
DJ and Brendan Broadbin came to our Hope in Focus LCA Family Conference with a lot of questions about their little boy’s blindness, and they left with amazing answers leading to innovative treatment for his type of Leber congenital amaurosis (LCA).
The couple traveled from their southwestern Connecticut home to the July 2019 Philadelphia conference knowing their son Jace had LCA, but not knowing the specific form of the rare disease because their 11-month-old had yet to be tested genetically.
Jace ready for the day
Jace’s parents introduced themselves to retinal specialist Prof. Michel Michaelides, one of Britain’s top ophthalmologists, a founding member of MeiraGTx, where he is Head of Clinical Ophthalmology, and a conference panelist.
“At that time,” DJ said, “we hadn’t even met with a geneticist yet, but Michel gave us his contact information ‘just to have.’ We then got to hear the panel discussion at the conference regarding the clinical trials that were taking place across several of the gene variants.
“A few days before we went to Boston to hear Jace’s genetic results (in October 2019, three months after the conference), we received the Sofia Sees Hope (now Hope in Focus) newsletter in the mail, outlining the treatments that were discussed at the conference.
“We brought the newsletter with us to our appointment and almost fell out of our seats when we learned that Jace had the AIPL1 variant, and that MeiraGTx was currently working on treatment through the Compassionate Use case program in the UK – we emailed Michel that same day.”
Compassionate Use treatment
Dr. Michaelides is a Professor of Ophthalmology at University College London Institute of Ophthalmology in the Department of Genetics. He also serves as a Consultant Ophthalmologist at Moorfields Eye Hospital in the departments of Inherited Eye Disease, Medical Retina, and Paediatric Ophthalmology.
The professor has discussed the United Kingdom’s Compassionate Use program in a Hope in Focus webinar series episode called “Let’s Chat About…Gene Therapy for LCA,” describing LCA4 as an exceedingly rare and severe form of the disease in which children have profoundly reduced vision from birth.
A special unlicensed medicine is one manufactured without marketing authorization from the Medicines and Healthcare products Regulatory Agency. The agency only grants a product license once a medicinal product has been proven to be safe and effective. Prescribed products not holding a marketing authorization include those prepared on an individual basis by “special order” manufacturers, according to the National Institutes of Health.
Floating jellyfish capture Jace’s attention
“There is a narrow window of opportunity (for treatment) because the retina degenerates and thins out by the age of 4 years,” Dr. Michaelides said. “Treatment needs to be before 4 years of age. MeiraGTx has manufactured a gene therapy that they are making available under a Specials license in the UK.”
After months of conversations, sharing test results, and talking with the hospital board members and surgeons, Jace received approval for the Compassionate Use treatment in one eye on March 17, 2020.
The family traveled to London, Jace underwent pre-op testing, and the surgery was cancelled: The world had just begun shutting down because of the COVID-19 pandemic.
DJ and Brendan thought the surgery would be cancelled indefinitely, but to their wonderment, they returned in September, quarantined for two weeks, and Jace received the gene therapy in his left eye, which is stronger, on Sept. 30, 2020. He had just turned 2 that August.
Jace after undergoing Compassionate Use gene therapy
Before LCA4 (AIPL1) surgery
The couple first began to realize when Jace was about 8 weeks old that he was not looking at them or trying to track toys.
“He was smiling from touch, but never in response to one of our smiles. He wasn’t blinking when lights were shown in his eyes and wasn’t shutting his eyes or even squinting in the brightest of sunlight. His eyes never seemed to move out of that ‘newborn’ stage of being all over the place,” DJ said.
“When we brought him to the pediatrician, hoping we were just being paranoid and this was something he’d grow out of, they confirmed that something wasn’t right and within an hour we were meeting with a pediatric ophthalmologist – kicking off a year-long journey for answers.”
Before his surgery, Jace had minimal light perception and not much functional vision.
“Lights had to be very bright for him to react to, and his reaction was at least two seconds delayed. Phone screens and TV screens were not bright enough to elicit a reaction from him and outside he had to always be in sunglasses because the sun was never too strong for him to look away from.
“With his left eye, we felt like he could have some shadow perception or make out very high contrast shapes and objects,” DJ said. “He had some words at the time, and labeled toys by feel and sound, but never by sight.”
Prof. Michel Michaelides
Post surgery
About a year after Jace’s surgery, his parents – now both 33, with mom working as a market researcher and dad as a sourcing manager for a major retailer’s store design team – welcomed another son. Jace’s little brother, Gio, just turned 1 in August.
Gio is sighted and in awe of his big brother – so much so, DJ and Brendan said he always felt like a toddler to them, missing the infant stage, because he’s always trying to keep up with Jace.
He learned how to crawl so he could be closer to his brother and now runs out of bed to meet Jace every morning,” DJ said. “Jace assumed the ‘patient older brother’ role incredibly well. He’s even learned to share his most favorite toys and it’s music to our ears when the boys are both belly-laughing as they rough-house with one another and try out their wrestling moves.”
Gio loves his big brother
Since the surgery, Jace can identify most of his toys by sight when they’re held three feet or closer to him.
“He is especially good at identifying the ones that are brightly colored and his favorite cars and dinosaurs, of course,” she said.
The couple believe Jace gained valuable functional vision from his surgery.
“Our hope was always that the surgery could protect some of the light perception he did have for a bit longer, never imagining that it could lead to anything more.”
“Someone who received LUXTURNA® described vision improvement as regaining ‘pockets’ of vision in the area where the retina remained intact, and this is exactly how we believe Jace has also regained some vision in his left eye. He will turn his head in certain directions to get a better look at what is in front of him.
Brendan, Jace, Gio, and DJ Broadbin
“In terms of being able to better navigate, Jace now bends down to pick up small objects that might be in his way, noticing them solely based on sight, not feel.”
Jace’s teachers have commented he’ll squat down to look under things when he wants a specific toy in the classroom and stand on tippy toes to find things placed on countertops.
“This makes us laugh to hear.”
Jace, now 4 years old, smiles when he catches a glimpse of his favorite people and things.
“It is heart melting. But he also still flashes that same perfect smile when he feels the sand at beach, hears his favorite country songs, or tastes an ice cream cone – so yes, his vision has changed and it’s amazing to experience, but to us he’s always been amazing to experience.”
P.S. Hope in Focus featured the Broadbin family in its annual Dinner in the Dark video in October 2022. Please click here to view.