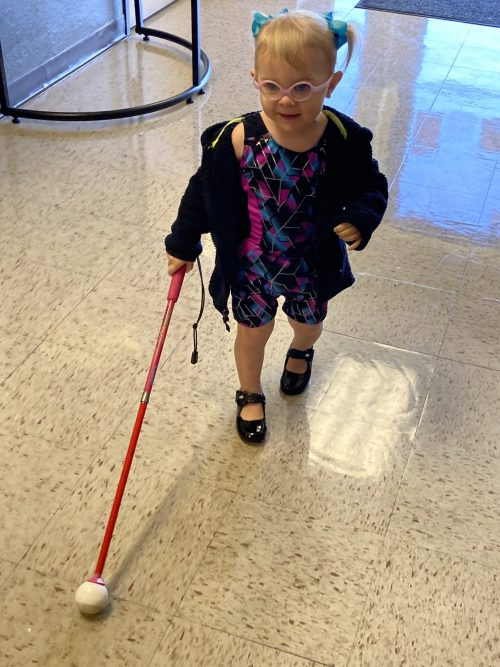
I am 22 years old and I have LCA.
My name is Angélica Bretón Morán, I am from Mexico, I am 22 years old and I have Leber congenital amaurosis (LCA). Two years ago I learned that the gene that affects me is the RPGRIP1.
I’ve been reading many sites on the Internet, and I have realized that there are many comments from parents concerned about the development of their children, however there are almost no comments from parents with older children or people with LCA who talk about their development.
My intention is to tell you a little about my story to bring you peace.
LCA diagnosis: Many wrong turns
When I was 2 months old, my mom realized that I did not follow people or toys with my eyes, and that when they did not speak, I cried as if I was alone and my eyes were standing down.
My mother is licensed in special education and specialized in hearing and language; she has many diplomas in different disabilities, including visual disability, so she realized that something was not completely normal.
The doctor told my parents that my eyes had to mature, but the months passed and nothing changed. My mom was certain that my characteristics were those of a blind person.
Finally, the doctors diagnosed that I was blind and there was a rain of bad diagnoses, syndromes that would terribly affect me, or that I would die a premature death, or that I would be like furniture, without the ability to do anything. They also said that maybe I would lose some other sense, that I could never talk, walk, eat ,among many other things Of course all this broke my parents’ hearts.
Other doctors recommended surgery but my mom never agreed and my dad supported her, he trusted in her because my dad did not have the experience with people with disabilities like my mom.
It was a painful road for my parents until a doctor told them “If they force me to give a diagnosis, I would say to you that it is LCA.” I was 2 years old.
My dad searched for information about this new diagnosis on the Internet, and what he found was not nice things, as I see that happens today. In one of the Internet searches, my parents found a writing made by a 22-year-old Italian young lady who was a musician; this for them was something hopeful.
Live with LCA: ‘I want to be a pianist’
From that moment we were in the group of research families, and for many years that was the diagnosis even though there was nothing that would formally confirm it. However, my parents did not lose details about my development; they were still worried because there was not something completely defined, since the disease was little known.
While my mom stimulated my touch with toys with different textures, placing hands in containers with different seeds, she strengthened my hands and enrolled me in mud classes. I was very afraid of the people, so she enrolled me in ballet classes where I would meet more girls and teachers. I was very little and I liked the music and the stage.
Then I went to kindergarten. It was not easy to find a school where they would admit me, but my parents never agreed that I was in a school for people with disabilities. As I had already been in the Ballet, it was easier for me to adapt to the classroom.
My parents never treated me differently, when I told my mom that when I grew up I wanted to be a pianist and opera singer, she told me that I could reach where I wanted. I liked to hear Charlotte Church and I admired Andrea Bocelli, because he was an example for me.
I have always been shy, I work hard to be sociable. This has been good, even though it has not been easy, but the reality is that we live in this world that will not be especially easy for us. This is achieved gradually and with personal life experiences, but I believe that above all with great patience and understanding of our parents and relatives, then we personally will learn to have it.
To give an example, my mother let my friends go to my house, and she let me go to their homes too. My parents did not make me see that my case was something special, because I could play with dolls, the kitchen, to be a doctor etc., like all the girls; only that when there were obvious things like playing jump on the stairs, the adults had to tell us that this was dangerous, because this is dangerous for any child, do not you think?
LCA, love and discipline
When I behaved differently from other children, my parents told me that this was not a good behavior. They explained to me how I should behave, without showing myself. When I waved my hands my mother held them and told me not to do it, she did it kindly but firmly and little by little I stopped doing it.
What made my parents angry was that I pressed my eyes with my fingers, because this was harmful to my eyes, every time they saw me doing it, they rebuked me more strongly; but now I thank them, because my eyes have no harm and in the future if the cure for my gene is found, there will be no injury that prevents me from receiving the treatment.
My parents educated me with a lot of love, but above all with discipline, an orderly and coherent discipline. I had to keep my toys like all children, learn to eat correctly like everyone else, not put my hands in food and just touch it with a finger that my mom called “guide finger”, this as a support because I cannot see the food.
I am weak visually, when I was a girl I could see better than now. Now I see lights and shadows but I cannot distinguish differences between colors as I used to, this has been so progressive that I did not realize until recently.
My disability has never been a secret, but neither has it been something for which I have to be different from others. It is true that you have to adapt some things, this is logical; however you have to look for how to achieve the objectives.
Going back to my life story, I have always studied in regular school; all my life music has been present. I graduated from music training at the Autonomous University of Nuevo Leon (UANL) while studying elementary school. I finished high school and got the second place for qualifications. I entered the technical career in music and finished it with honors while I studied online high school, all this in the UANL. I am currently studying a degree in music with an emphasis in piano at the same university and I have been studying professional singing for five years with a private teacher to become an opera singer.
Brother and sister
I think it’s important to mention that I’m not an only child, I’m the oldest of two. My parents, like many of you, were afraid that the second child would have something worse than mine, but they have always had great faith in God and my brother was born. He does not have any disability, but he was asthmatic since he was a child and my parents have faced their situation in the same way they have faced mine … they never treated him differently, nor did they make him see that he had disadvantages. But to achieve something you had to look for how to do it. He is a high-performance swimmer, asthma attacks are becoming less frequent, and he is currently a healthy boy.
All this I tell you so that you know how important it is not to limit yourself. If you limit yourself as parents, you will limit us as children. My brother has been a great support for me in many situations, he has taught me to be the older sister. Our illnesses and the education of my parents, they taught us that neither he nor I have an obligation to take care of each other, we simply do it because we are brother and sister and we love each other
I played with my brother, we made mischief, we laughed late at night until my parents scolded us to sleep, as any pair of siblings can. My parents have never marked differences between us, everyone is accepted with their differences, we all have, but I am not more important than him, nor is he more important than me.
All this we have worked for 23 years as a family, we have been wrong many times, some other times we have felt that our effort has been in vain. We have stumbled like any family, we have asked for help when we consider that it is necessary. We have also tried to help when we think it is appropriate. But above all things, the strength of my parents and my family has been their faith in God, which they have transmitted to me and my brother and this faith has been my reason to continue in difficult times.
Speaking about people with LCA, they may have other disabilities, I have witnessed them myself, or they may not have them as is my case. However, I can tell you that when you treat a person different from the others, that person will behave differently regardless of whether they have a disability or not.
Live with LCA: Find your way
You have to be aware of the limitations, but you also have to find a way around the obstacles, or pass through them, or use them as a catapult, or see them as a feature that makes us unique and special as people, as we all are.
I hope my words help many families, since that is my intention. I invite more adults with LCA to tell their stories and how they have faced life! I think we would give a much more encouraging approach when new parents enter the Internet looking for information about the diagnosis of their children, and why not, for those who are newly diagnosed patients and research the internet on their own.
I have known fathers and mothers with LCA, young people, babies, children, even a child who was cured with genetic therapy because he has RPE65. We met him when he was blind and the second time we saw him, he saw and guided the other children who were still blind. It was amazing! We were all very excited and we could not stop the tears.
Finally I want you to know that I am in the best position to answer your questions in this post from my personal perspective. As I mentioned before, my only intention is to help because I am greatly moved to see your anguish and I feel that it is my duty to be now the 22-year-old girl who brings a hopeful message to those who enter the internet looking for information about LCA.
I would like to know, is there anyone else that has the RPGRIP1 GEN? I do not know anyone else!
God be with each one of you, blessings!