This is the fifth in a series following the progress of Creed Pettit, a 9-year-old Florida third-grader, who completed treatment in March with the breakthrough gene-therapy drug called LUXTURNA™, approved as the first gene therapy for RPE65 genetic mutations and as the first-ever genetic therapy in the United States for an inherited disease.
Life looks a lot brighter for Creed Pettit, the Florida boy who just became one of the first LCA-RPE65 patients to have eye surgery using the brand-new gene therapy treatment LUXTURNA™.
For the very first time, Creed built the entire track of his wooden train set, and he did it in the garage rather than in the house with all the lights on.
“He played with it for over two hours,” said his mom, Sarah St. Pierre Pettit. “He played with it like a kid should play with it.”
Before the surgery, Creed and his mom would have what she called “taught play,” with her little boy doing only what he was taught or told.
“All of a sudden, he was playing with these things, interactive play, putting doors on the train, making the bridges of the train set.”
Even digging into the box with the train-set pieces is a new experience. Before, Creed wouldn’t look in the box because it was dark, and he couldn’t see anything. Now, he’s looking into the box and pulling out curved pieces of track.
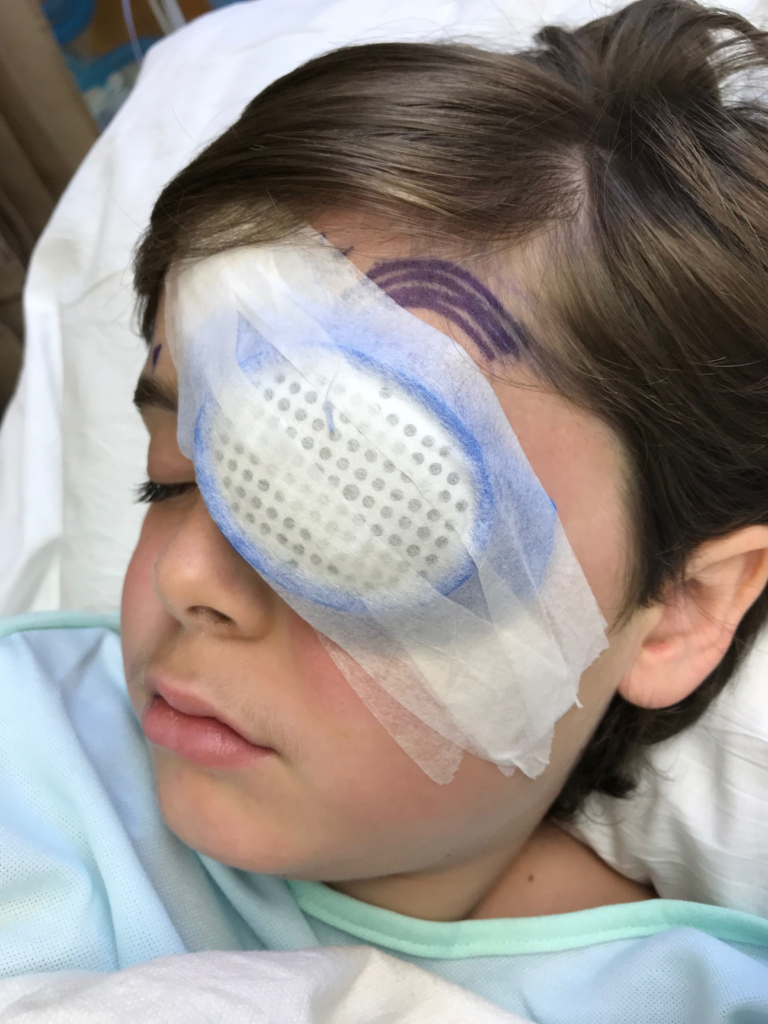
Creed is back home in Mount Dora, Fla., from having his second surgery, this time on his left eye, on March 28 at Bascom Palmer Eye Institute in Miami.
He and his family came home to a house decorated by friends with streamers, balloons and a big home-made sign that said, “Welcome Home Creed.”
Creed is one of a handful of the first LCA-RPE65 patients to receive the genetic therapy treatment LUXTURNA™ developed by Spark Therapeutics.
The U.S. Food and Drug Administration granted approval of the revolutionary drug in December. Soon after, preparations began for administering the treatment to the first patients the week of March 19. The surgery entails injecting under the retina a human-engineered virus containing copies of a normal gene, prompting the making of more RPE65.
As Creed came into the operating room last week, he heard “I’ll Be There For You,” coming from the speakers; it’s the theme song from the television show “Friends,” and the same song Creed and his mom – and the whole surgical team – sang while waiting out a short delay before his first eye surgery.
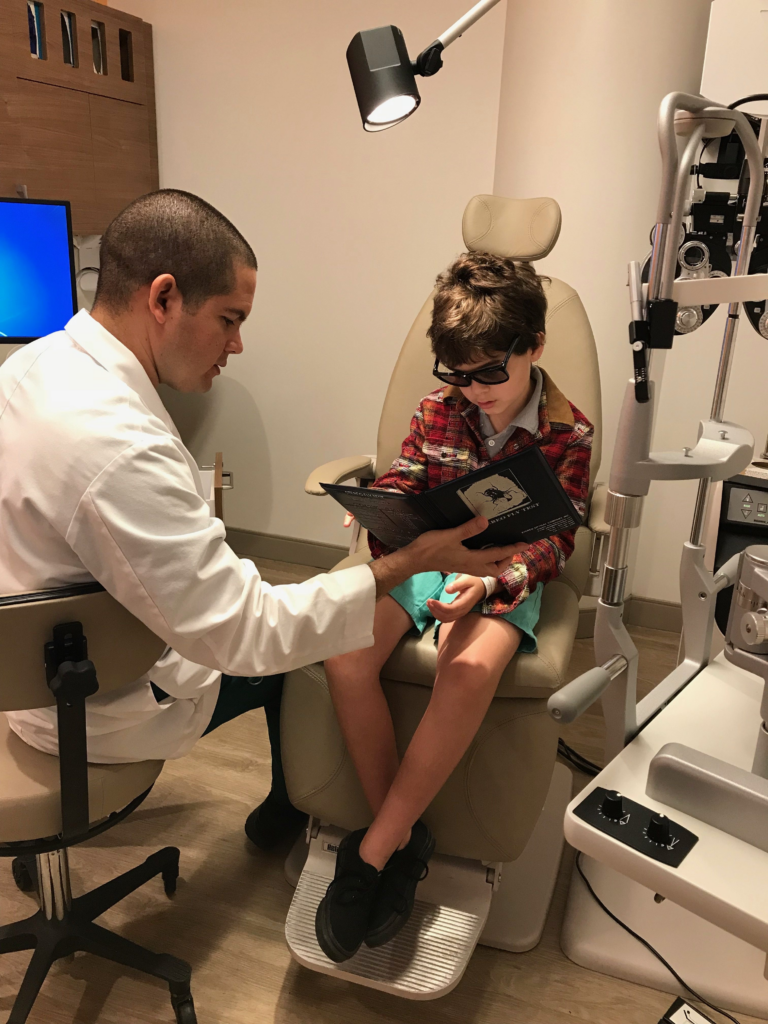
After performing surgery on his right eye on March 21, Dr. Audina M. Berracol found new growth in the eye’s photoreceptor. Creed will return to Miami on Monday for high-resolution scanning, called Optical Coherence Tomography, (OCT) on his left eye.
Back at home using just basic lighting, Creed reads “Diary of a Wimpy Kid.” And he looks down at his food now.
“It’s really, really neat,” Sarah said. “Every day has just shocked me.”
He watched “Peanuts,” one of his favorite shows, and then went on a long walk.
“It’s just so cool to watch him. He has his sunglasses on, he’ll just stare, looking at different trees, touching them.”
No more flashlights, either. Years ago, Creed’s mom outfitted their home with bright lights and lots of flashlights, so he could see things if they dropped under the table.
The third-grader also stopped by his school to see his friends and he kept looking at the designs on the floor and asking people what they were eating.
“It’s a hamburger,” his mom said. “It’s almost like teaching a toddler all these things.”
Speaking of school, after Creed returns from Miami for his follow-up appointment on Monday, he goes back to school Tuesday and plans to wear a new T-shirt, much to his mother’s chagrin.
Sarah wanted to buy Creed something special to celebrate the success of the surgeries and his new-found look on life. She wanted to buy him a watch or something cool at one of the popular stores.
“Look, here’s a neon one,” she said, pointing to a big watch. “Anything you want.”
Instead, he picked out a shirt with words on it, “totally not something I’d ever buy,” Sarah said. The shirt says, “I was going to do my homework, but my hands are full,” and shows an image of two hands holding a game console.
She thinks he picked it out because he could read the letters.
“He’s never gotten in trouble (at school). He thought it was hilarious that it said that.”
Sarah said she was thankful that her mom and her fiancée accompanied her and Creed to Miami and helped keep her calm after waiting nine years for Creed’s treatment come to fruition.
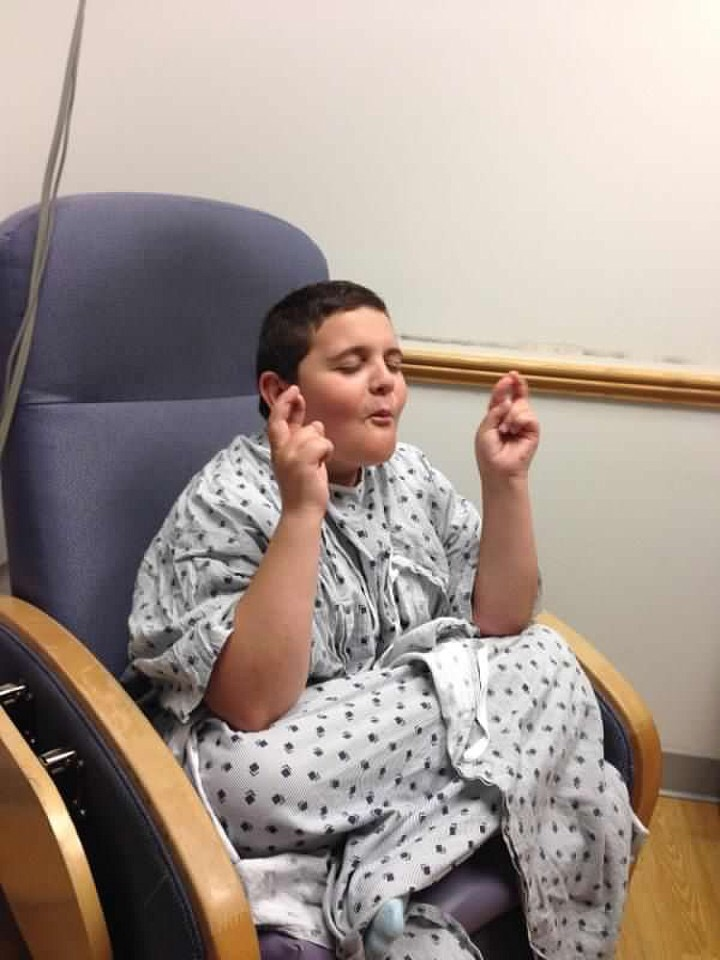
“I think I cried harder on the second one,” Sarah said after this latest surgery.
“Oh my gosh! It’s done! We’re done! We beat this! We never stopped fighting and look where we are now.”