After crazy months of looking for answers to questions about her infant’s vision, Melissa Matias learned her baby girl, Dylan, had a form of Leber congenital amaurosis known as LCA6 caused by mutations in her RPGRIP1 gene, a protein needed for healthy photoreceptors.
“I put my big-girl pants on and said, ‘This is the card she and I have been handed.’ ” The Georgetown, Texas, mom added: “There’s a reason for this.”
When Dylan received her confirmed genetic diagnosis in May 2020 at age 5 months, her mother did not assume someone else had the time or resources to search for a treatment or cure for her daughter’s form of blindness. Melissa organized people, advocates, and information. She created the RPGRIP1.com website and began collaborating with the nonprofit biotech Odylia Therapeutics on a $4 million fundraising effort to advance therapy for the RPGRIP1 LCA6 program.
The Atlanta-based biotechnology company is in late-stage preclinical trials, with the goal to be in clinical trials in about two years. Melissa is hoping Dylan – a gregarious trampoline-jumping gymnast – will benefit from this developing gene therapy.

“We aren’t trying to fix or change Dylan. There’s nothing wrong with her. But if there’s a chance for gene therapy to enhance her life, you would want to do that,” she said. “Blindness doesn’t mean darkness. It means ‘I can do this; I just might need to do it in a different way.’ ”
Dylan’s doctor told the Matias family that the possibility of sight-saving therapy coming to fruition was not a matter of if, but when.
“This therapy, if it’s in reach, let’s get going,” Melissa said. “Let’s get the word out and open the door to gene therapy, not just for RPGRIP1, but for other genetic visual impairments and rare diseases.”
Financing poses the major obstacle. Melissa enlisted the help of patient families and organizations, such as Hope in Focus, to help spread the word and secure necessary funding to bring the treatment to market. About $115,000 has been raised, including donations from Dylan’s brothers, 8-year-old Colton and 12-year-old Brayden.
Funded by Massachusetts Eye and Ear and the Usher 2020 Foundation, Odylia Therapeutics started in 2017 with the goal of facilitating treatments for low-prevalence retinal disease with proof-of-concept science to Phase I/II/III clinical trials.
LCA6/RPGRIP1 Gene Therapy – Following LUXTURNA® Path
The gene therapy uses vector technology developed by Odylia Co-founder Luk Vandenberghe, PhD, Director of the Grousbeck Gene Therapy Center at Mass Eye and Ear, and Assistant Professor of Ophthalmology at Harvard Medical School. The research builds on proof-of-concept data generated at Mass Eye in the labs of Vandenberghe and Eric Pierce, MD, PhD, a physician and surgeon at Mass Eye and the William F. Chatlos Professor of Ophthalmology at Harvard Medical School.
LUXTURNA® currently is the only gene therapy on the market. Developed by Spark Therapeutics and federally approved in 2017, the drug treats a form of LCA known as LCA2 (RPE65), and it is the only gene therapy approved in the United States for any inherited disease.
Odylia hopes to follow in LUXTURNA’s path with its research. Delivered by injection under the retina in each eye, LUXTURNA® has helped improve vision in patients for the last four years.
“The success of LUXTURNA has proven to be life changing for many patients,” Odylia said in a news release. “It’s very exciting that with the efforts of Odylia, those affected with RPGRIP1 mutation could possibly have the same opportunity in the near future.”
Developing treatments and administering them to young patients is paramount to stall vision deterioration, as LCA6, along with the more than 27 forms of LCA, is degenerative.
Odylia says its goal is to bring proven therapeutics to patients, regardless of the number of people with the disease or the opportunity for a company to make money.
By identifying promising treatments, the company said it can partner with patient groups, community experts, and financial sponsors to develop strategic, scientific, and clinical plans. Through collaboration with industry and foundations, the organization said it can move treatments from concept through development and to patients with rare diseases.
These partnerships are needed for the estimated $3.9 to $4.3 million in development costs to advance the research through toxicology studies and fund clinical manufacturing costs. Odylia says it will work to lower costs and reduce overall expenses.
Partnerships and funding also will help with Odylia submitting an Investigative New Drug (IND) application to the U.S. Food and Drug Administration. Federal law requires that a drug be the subject of an approved marketing application before it is transported or distributed across state lines. Because a sponsor will probably want to ship the investigational drug to clinical investigators in many states, it must seek an exemption from that legal requirement. The IND is the means through which the sponsor technically obtains this exemption from the FDA.

LCA6/RPGRIP1 Research Receives FDA Designations
The FDA recently granted both orphan drug and rare pediatric disease designations for Odylia’s lead gene therapy. The company said the designations are granted for the “treatment of RPGRIP1 mutation-associated retinal dystrophies,” which most commonly includes LCA6 but is also associated with diagnoses of cone-rod dystrophy 13 (CORD13) and forms of early-onset retinitis pigmentosa (RP).
“Receiving orphan drug and rare pediatric disease designations represents an important milestone for Odylia and recognizes the potential of this gene therapy to deliver life-changing results to LCA6 patients and RPGRIP1 associated retinal dystrophies,” according to Ashley Winslow, PhD, Odylia’s Chief Scientific Officer.
Between 400 and 600 people are affected with LCA6 in the U.S., with about 20,000 globally.
Orphan drug designation is granted to therapeutics intended for treatment, diagnosis, or prevention of diseases affecting fewer than 200,000 people in the United States. Rare pediatric disease designation is granted to serious or life-threatening rare diseases that primarily affect individuals under age 18. Orphan drug and rare pediatric designations provide companies like Odylia with benefits, including access to research grants to support clinical studies, waiver of regulatory fees, seven-year marketing exclusivity, and eligibility for a priority review voucher.

Struggle to Find Confirmed RPGRIP1 Diagnosis
Melissa’s persistence pushed her through incredibly trying times, having six back-to-back miscarriages during 2016 and 2017 after having her two sons. Two years later, she and her husband, James, got the news she was pregnant with Dylan.
“She was so meant to be,” Melissa said. “It was unreal when it came time to deliver. Everyone in that room was crying.
“They had all seen on my chart that I had 10 pregnancies. They were all just like…” she said trailing off. “It was such an emotional time.”
Two months later, though, Melissa and James realized something didn’t seem right with Dylan’s vision; her eyes jerked from side to side from a condition known as nystagmus. It was February 2020 with the tentacles of COVID spreading over the globe, creating an overworked, overwhelmed medical world focusing on patients infected with this strange new virus.
Finding an initial diagnosis for Dylan proved extremely difficult, especially when one doctor said nothing was wrong, probably just delayed maturation of vision, and another mentioned LCA but brushed it off. So, Melissa did what we all do now when we don’t get answers: She Googled the disease.
“The more I learned about LCA, I knew in my heart right away what we were looking at. But I’d also think, it’s so rare, what are the chances?”
She also learned the next step to move forward with treatment or care for 2-month-old Dylan depended on a confirmed genetic diagnosis determined through genetic testing.
No genetic testing, not now, she was told. Not until COVID is over. Melissa wasn’t waiting for the end of the pandemic, now into its third year. She knew she needed a doctor’s order but hadn’t yet connected with a retinal specialist; she sought out the help of Dylan’s pediatrician, a doctor she characterized as amazing, who ordered the test.
Waiting for the results, Dylan’s parents scheduled an appointment for May 28 with a Houston retinal specialist; the confirmed diagnosis of LCA6/RPGRIP1 came through May 27, paving the way to finding resources in orientation, mobility, and education for Dylan.
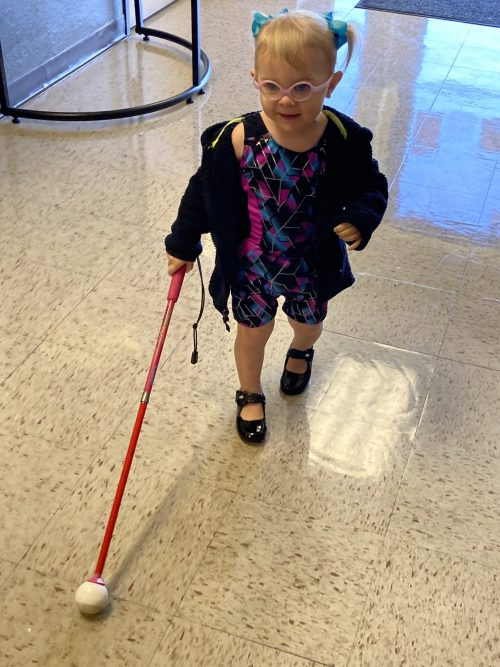
Living with LCA6/RPGRIP1 / Like Mother, Like Daughter: Stubborn and Persistent
Dylan is now 2 years old. Her parents believe she has tunnel vision and does best seeing objects 10 or 15 feet away. She’s better now at keeping her glasses on, rather than ripping them off all the time like she used to.
On her first day of Orientation and Mobility training, she picked up a cane and used it just the right way, a milestone the instructor said he had never seen in a child her age.
“Indoors, she’s just amazing,” Melissa said. “She loves picture flashcards, playing in her toy kitchen, always wants someone to read her books, and she loves her new mini trampoline she got for Christmas. And music – she loves to sing and dance.”
Dylan takes her time in new environments and is a little more cautious than other toddlers because of her visual limitations. With photophobia being one of the symptoms of LCA, Melissa said her daughter finds bright outdoor sunlight to be the most challenging and where she relies more on her cane skills.
The little girl also is very vocal and visual.
“Her vocabulary is comparable to a child much older than 2,” Dylan’s mom said. “We know she is very visual, something we feel blessed with, since LCA can present with a wide range of visual capabilities. We’ve actually had to teach her to bend down and feel for objects she drops, because her instinct is always to look for something first.”
She’s also persistent.
“She has a very strong desire for mastery, having to do things over and over again. She likes a challenge.
“I have no doubt this girl is going to change the world. No matter what happens if gene therapy comes about for her or not. She’s going to be on skis this winter. I’m not going to give her an excuse not to do something or at least to not try.
“She can and will do anything she wants, just in her own ways. Stubborn and persistent, exactly what she needs.”












{kind=link}